Clinically important drugs acting on sodium and calcium channels
by egpat 30-08-2017
Drugs can act on various targets to produce pharmacological response among which ion channels have their own significance. A drug can act on ion channels in two ways. First, it can directly block the ion channel by producing a physical block with the ion channel so that ion transport across the membrane can be prevented. Second, it can act at an accessory site to modulate the ionic channel activity and may either increase or decrease the opening of ion channels hence called as modulator. Most of the drugs act as negative modulators on ion channels.
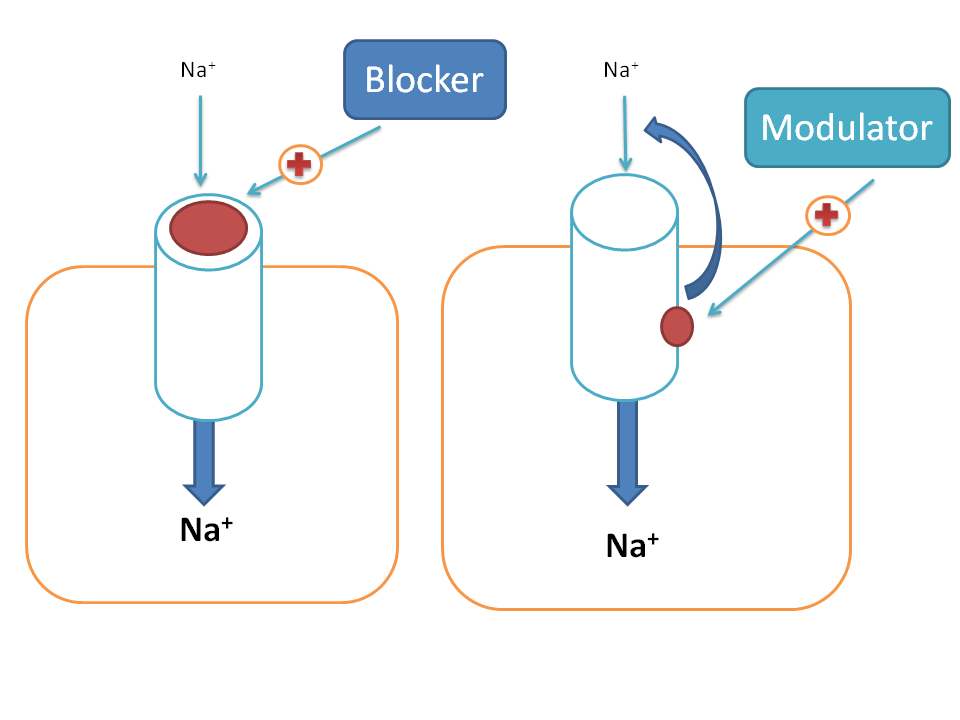
In our physiological system we come across with so many ion channels such as sodium, calcium, potassium, magnesium and chloride channels. All these may not serve equally as good drug targets and here we will discuss few of the clinically important drugs that act on any of these ion channels.
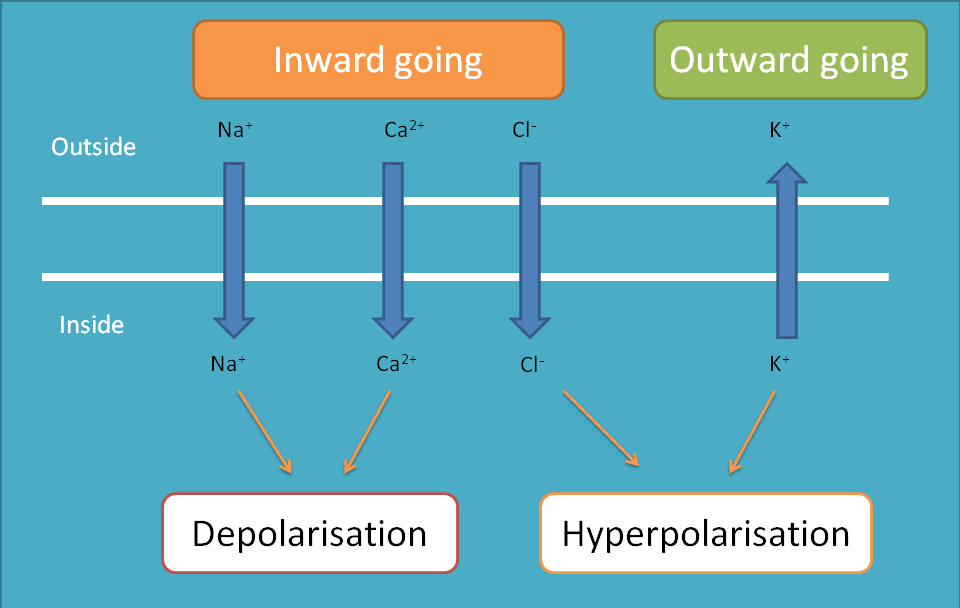
Sodium channels
First we will start with sodium channels.
What is there role of sodium channels in our physiological system?
As it is well familiar that sodium channels are always inward going, that is, they always enter into the cell making depolarisation of the cell. So, within a cell these channels produce depolarisation and between the cells they produce impulse conduction.
That’s fine. Now our next question, how these sodium channels are controlled? Which factor opens and closes these ion channels?
We are coming to the point. These ion channels activity can be controlled by various gating mechanism of which three are discussed below.
- voltage-gated
- Ligand-gated
- Concentration gradient gated
We will go with one by one along with thier location, role and drug targets.
Voltage gated sodium channels
So, let’s start with voltage gated sodium channels. These channels are opened when the cell receives a signal in the form of action potential. So, when a cell receives action potential, the membrane potential slowly raises from resting membrane potential to threshold potential where these sodium channels are opened.
Let’s consider the example of cardiac cells. Normal average resting membrane potential of cardiac non-pacemaker cell is around -90 mV to -80 mV. When action potential reaches to the cell through gap junctions, the membrane potential rapidly raises to the threshold potential such as -70 mV where fast acting voltage gated sodium channels are opened leading to a sudden raise of membrane potential to the positive side.
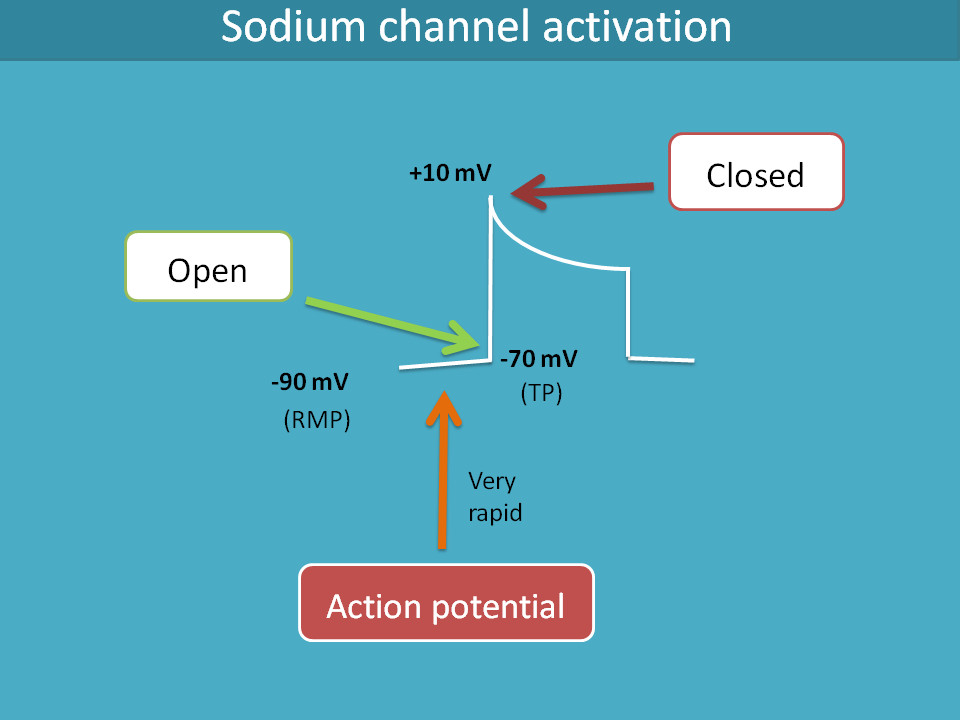
The same gating mechanism which opens the sodium channels also plays its role in their closing. When the membrane potential reaches to +10 mV, the sodium channels are closed.
In this way, these sodium channels are opened and closed by the membrane potential hence these are called voltage gated.
What is their location?
Voltage gated sodium channels present at various locations and among them three location are important in view of drug action.
- Heart
- Central neurons
- Local neurons
Heart
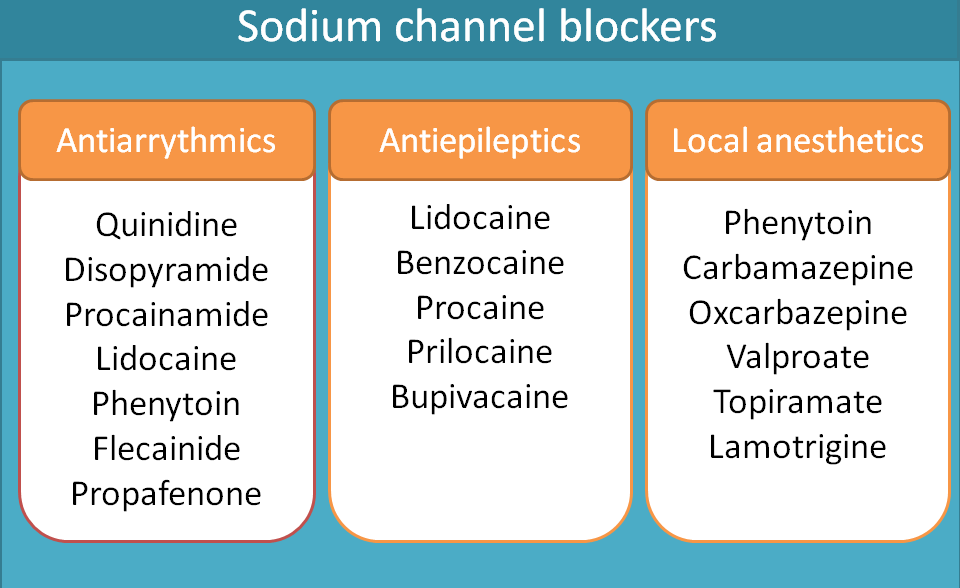
On the heart, the role of these sodium channels is to produce rapid depolarisation during the phase 0 of the action potential of the heart. Class I antiarrhythmic agents block these voltage gated sodium channels and slow the rate of depolarisation.
Class I antiarrhythmics can be sub classified into three categories such as class Ia, Ib and Ic. Various drugs included in these categories are given below.
- Class Ia
- Quinidine
- Disopyramide
- Procainamide
- Class Ib
- Lidocaine
- Phenytoin
- Mexiletine
- Class Ic
- Flecainide
- Propafenone
For complete classification of antiarrhythmic agents, see the below link.
Why they are sub classified. Are they not equal?
Yes, they are not equal in view of their kinetics of action. That means, they bind and release form sodium channels at different rates. We can categorise their interaction with sodium channels as follows.

Class Ic agents attach and detach very slowly with sodium channels whereas class Ib agents attach and detach rapidly within the time frame of the action potential. Class Ia agents fall in between these as they have intermediated rate of attachement and detachment with sodium channels.
Class Ia agents also block voltage gated potassium channels thereby extend action potential duration.
Central neurons
Sodium channels play a vital role in neuronal transmission within the CNS. They are responsible for fast neuronal conduction and CNS stimulation. But in few pathological conditions, they may be excessively and asynchronously fired leading excessive neuronal discharge resulting in epilepsy.
Now you can easily guess what the role of drugs blocking these channels is. Yes, you are right. Drugs acting on these channels are used antiepileptics.
A number of drugs fall in this category including phenytoin, carbamazepine,oxcarbazepine, valproate, lamotrigine and topiramate.
Note: Phenytoin is classified as class Ib antiarrhythmic agent as well as antiepileptics. Due to its adverse effects and better alternative drugs available, nowadays its use is confined to epilepsy.
Drugs like valproate, lamotrigine and topiramate are new generation of antiepileptics that act by several mechanisms among which one mechanism is sodium channel block.
Local neurons
Now the turn comes to the local neurons. Here again the role of sodium channels is same, that is to produce neurotransmission but locally. So, when these sodium channels are blocked, local neurons at the particular area can’t be depolarised and activated preventing transmission of impulse from periphery to CNS.
Drugs like lidocaine, procaine, benzocaine and bupivacaine act as local anaesthetics. Local anaesthetics can be further classified as ester and amide derivatives. For complete classification of local anaesthetics, visit the following link.
In this way, drugs acting on voltage gated sodium channels act as antiarrhythmics, antiepileptics and local anaesthetics.
Ligand gated sodium channels
Now, let’s see another type i.e. ligand gated sodium channels. These channels are coupled with receptors on which a suitable ligand binds to activate their opening. This type of ion channels play important role in communicating between the cells, particularly in the neurons, and responsible for excitatory post synaptic potential (EPSP). We can find such type of ion channels abundant within the CNS responsible for both fast EPSP and slow EPSP.
For example, AMPA receptors when activated by glutamate produces fast EPSP and NMDA receptors produce slow EPSP
As we have already discussed, these ion channels can be opened when a ligand binds to the receptor site. AMPA, NMDA and kainite receptors are coupled with sodium channels and when glutamate binds to these receptors, sodium channels are opened leading to depolarisation.
Drugs like phencyclidine, ketamine and dizocilpine block sodium channels associated with NMDA receptors.
Late phase sodium channels
In the cardiac cells, a small portion of sodium channels are opened at late phase after the depolarisation. Under normal conditions, they don’t produce any effect but in pathological conditions this may increase intracellular sodium levels which then increase intracellular calcium levels.
Cardiac cells can get oxygen supply during diastole which is impaired by this excess calcium leading to deficiency in cardiac oxygen supply and precipitation of angina.
Ranolazine is one of the drugs that blocks these late phase sodium ion channels hence indicated for chronic stable angina.
Renal tubular sodium channels
Till now we have discussed sodium channels gated by either voltage or ligand. But we can also find few of the sodium channels gated by concentration of ions at renal tubules.
As sodium is one of the essential electrolytes in the body, nephron always tries to absorb sodium from the filtrate back into the systemic circulation. But at collecting tubules, the last part of the nephron, sodium ions can’t cross the renal tubular membrane easily and require some specialised channels through which they can be absorbed. Here renal tubular sodium channels come into play. Renal tubular sodium channels are operated by concentration gradient of sodium ions and absorb the sodium from the filtrate back into the systemic circulation.
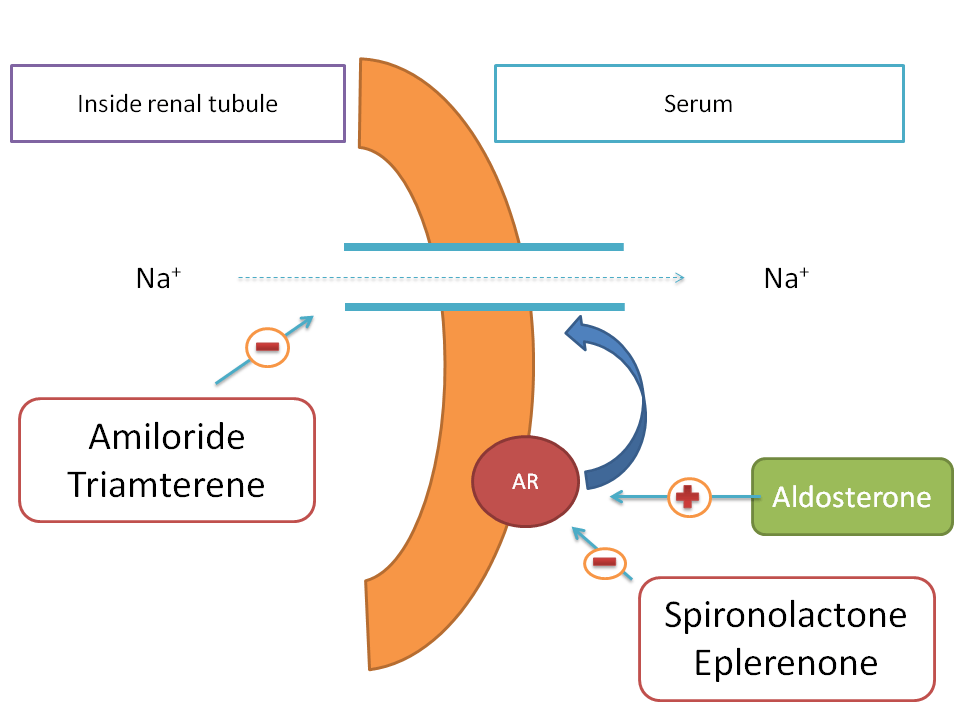
In few of the pathological condition such as heart failure, the increased sodium levels in the body produce more body volume hence more cardiac work. So, one of the approach in the treatment of the heart failure is to decrease the sodium levels in the body by using a diuretic.
Drugs like amiloride and triamterene block renal tubular sodium channels thereby decrease sodium reabsorption in to the body.
Aldosterone is a ntural hormone that acts on aldosterone receptor and increases the expression of renal tubular sodium channels. Hence it acts as positive modulator. Drugs such as spironolactone and eplerenone block these aldosterone receptors and thereby decrease sodium channel expression therefore act as negative modulators.
Calcium channels
Calcium channels are inward-going and when they enter into cell they cause depolarisation and most of the times contraction. Just like sodium channels, calcium channels are also gated by various mechanisms of which we will discuss few with important drug targets.
Voltage gated calcium channels
As similar with sodium channels, voltage gated calcium channels present at three locations.
- Heart
- Smooth muscle
- Central neurons
Heart
Cardiac action potential involves all ions like sodium, calcium and potassium. Calcium channels come into the picture at phase II of the action of potential. Phase II, also called as plateau phase, is maintained by calcium and potassium where the former is inward-going and later is outward-going.
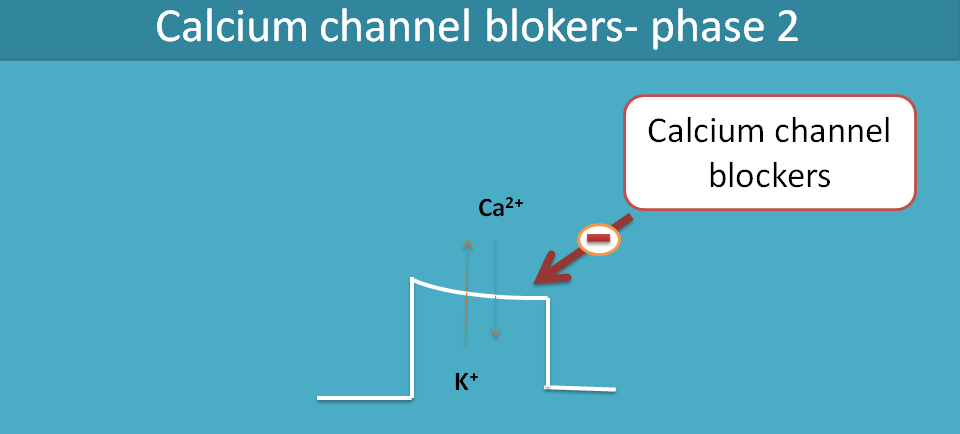
The net charge going inside is equal to net charge going outside so that these two ions maintain membrane potential remain constant during this plateau phase.
Abnormal increase in intracellular calcium levels [Ca2+]i may cause cardiac arrhythmias by delayed after depolarisation or even increase cardiac work and precipitate angina. Hence drugs like verapamil and diltiazem block these calcium channels by acting on the phase II of action potential of the heart.
Smooth muscle
The role of calcium in smooth muscle is obviously constriction. So drugs acting on these calcium channels act as direct vasodilators. Dihydropyridines sucha as nifedipine, amlodipine, felodipine, isradipine, nicardipine and nimodipine directly block calciu channels at vascular smooth muscle and hence indicated as antihypertensives.
Central neurons
In the periphery the main role of sodium is depolarisation and that of calcium is contraction. When the situation comes to the CNS, both sodium and calcium are responsible for neuronal conduction. Calcium mediated neuronal conduction is important at thalamocortical tracts and if this neuronal discharge is excessively fired due to enhanced activation of calcium channels, it may lead to absence seizures.
Now, a question may peep into your mind.
Calcium channels produce contraction in muscle but conduction in central neurons. How it’s possible? Are these channels same or different?
Exactly what you expect is true. Even both are calcium channels but they are different in type and functionality.
Those calcium channels present on cardiac and smooth muscles are of L-type calcium channels whereas calcium channels present on central neurons are of T-type.
One of the drug acting on these T-type calcium channels is ethosuxemide indicated for absence seizures.
That’s about the drug targets acting on sodium and calcium channels. We will cover drug targets acting on other channels in the next article. Your comments are welcome and share this post if you like.