Role of NMR chemical shifts in interpretation of spectra
by egpat 22-11-2017
Can we measure the absolute values of shielding around each proton in NMR? Obviously the answer is ‘No’. We can’t accurately measure shielding constant of every proton instead we can compare. Yes, here the concept of NMR chemical shifts arises to solve this problem.
So all the signals NMR are relative measurements comparing with a reference and how extent the protons are shielded is compared with the reference.
Then a new issue, which compound to be selected as reference? How it can be differentiated from the peaks of test sample?
We should think of it. Selection is tough. The reference, in an ideal case, should be pure, stable and easily available.
That’s enough? It should give a strong signal and to the best a single signal so that we can easily differentiate it from peaks of test sample.
Nice. So let’s pick one such compound. But where it should fall in NMR spectra, left or right? That means whether it should have high shielding or deshielding?
As we are always ready to face any challenge, so let’s take a reference with high shielding. So let the protons crowded with surrounding electrons.
Finally, we got the solution.
Tetramethyl silane, commonly called as TMS, is such a compound with 12 protons all equivalent and therefore giving a strong single signal.
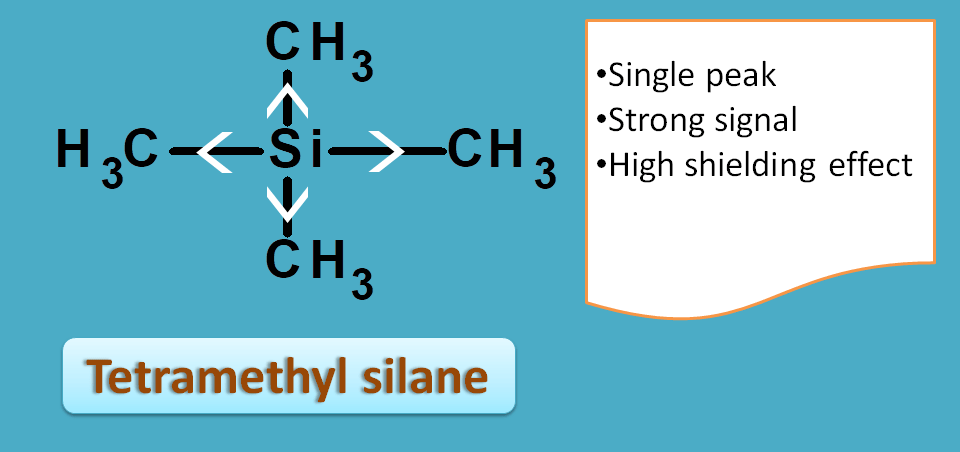
Due to the electropositive nature of silicon, all the protons are packed with electrons producing high shielding effect.
So peaks of test sample are measured relative to reference peak in NMR.
Let’s first understand the NMR chemical shift and then we can see how it works in interpretation.
What is chemical shift?
As we have selected a reference with high shielding effect it always falls to the right side of the spectra and most of the test peaks fall to the left side downfield in NMR spectra.
So, now we can define the NMR chemical shift.
In a simple way, it is the relative shift of peaks to the downfield from the reference peak in NMR spectra at a particular operating frequency of NMR spectrometer.
It is measured in parts per million and the scale is designed from 0 to 12 in proton NMR spectra and 0 to 200 in carbon NMR spectra.
Who is at 0 ?
No doubt, it’s our hero TMS. As it has highest shielding, it was given chemical shift value as zero.
By this time, we can clearly observe that nuclei with low shielding are farer from TMS peak. So,
Higher the shielding lower the chemical shift
That’s fine. But one question may peep into our mind. How this chemical shift is useful in structural interpretation in NMR?
Yes we are coming to that point.
Suppose we have a proton with similar environment in two different compounds. Let’s take butanol and pentanol.
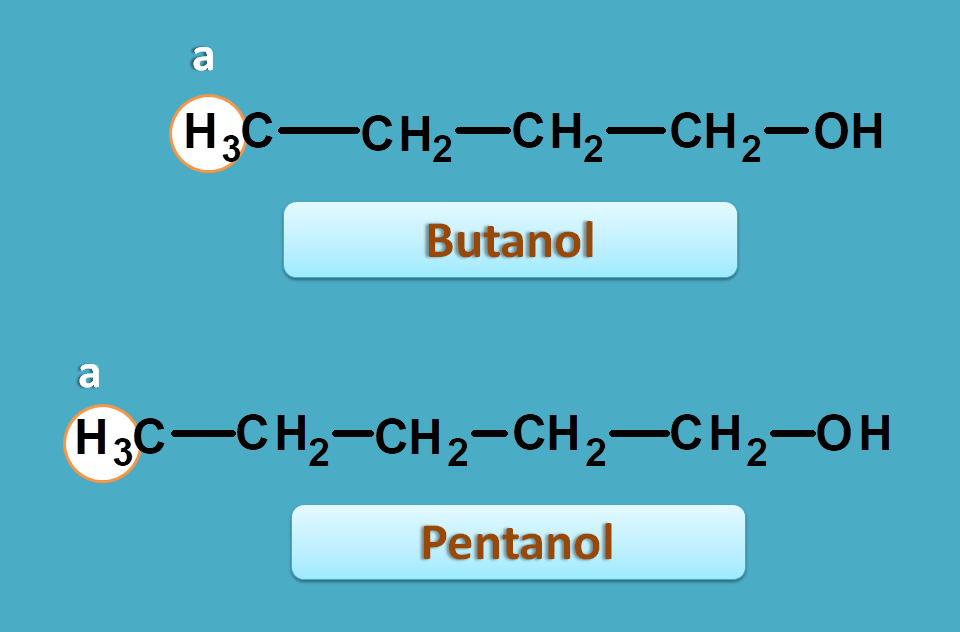
Now if observe proton NMR spectra of above two compounds, the methyl protons in both the samples will fall at approximate chemical shift of around 0.9 ppm.
That means both protons show the similar NMR chemical shift as they have similar chemical environment.
That’s great. So the chemical shift mainly depends on its immediate environment rather than on which skeleton it is present. Hence all the methyl protons with similar environment produce same chemical shift.
This makes chemical shift as best parameter to interpret the structure. We can have a NMR spectrum table compiled with approximate values of protons with different environment.
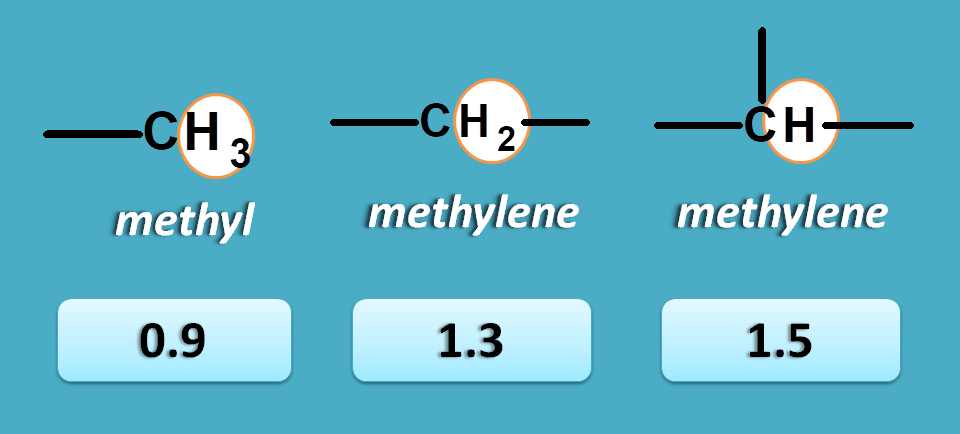
For example, methyl, methylene and methine protons can have approximate NMR chemical shift values as 0.9, 1.3 and 1.5 respectively.
You should remember that these values are approximate and may little vary with presence of other groups in the structure.
For example, the methyl protons in propanal may fall at 1.0 to 1.1 ppm that is practically very small in variation and can be easily interpreted as peak of –CH3.
In this way, we can predict the groups by comparing measured values with the NMR chemical shifts in the NMR table.
Okay, is that only use of chemical shift in NMR?
No, we can extend this concept for predicting the NMR spectra just by seeing the structure and comparing different groups. We can guess which proton may have high chemical shift before proceeding to NMR spectra.
Let’s have a look on the following example.

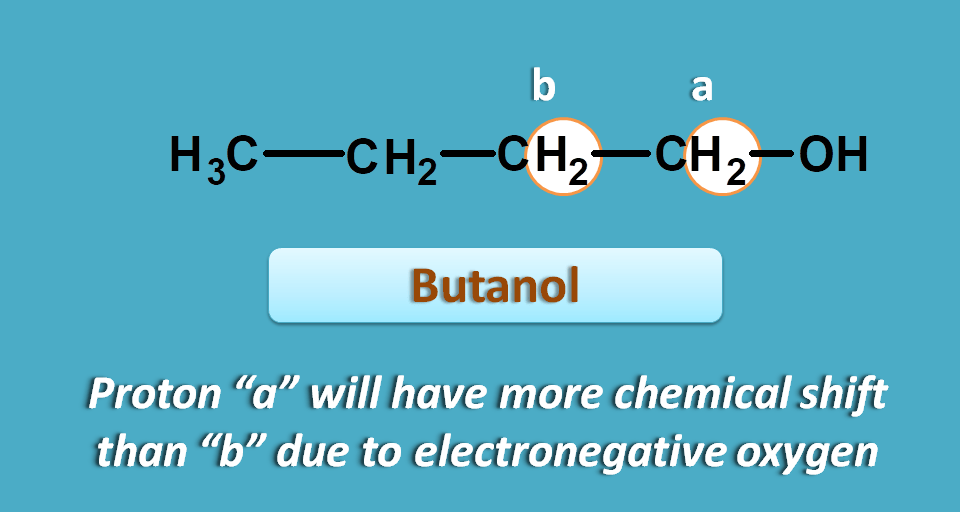
Here methylene protons indicated by “a” have different environment hence not equivalent in two compounds.
Due to presence of chlorine atom, which is highly electronegative, the electrons are pulled out of the carbon towards chlorine.
Now the carbon plays same role pulling the electrons farer from methylene protons proving a deshielding effect.
As we know that chemical shift decreases with shielding or in another word increases with deshielding, these protons show high chemical shift than normal methylene protons.
So, without going for real NMR spectra we can guess what happens to the chemical shift by the presence of adjacent groups.
To dig in to the more depth, let’s discuss three important factors influencing the values of NMR chemical shifts.
Factors affecting chemical shift
Three important factors can affect the chemical shift
- Electronegativity
- Magnetic anisotropy
- Hydrogen bonding
Electronegativity
As we have discussed above, the presence of electronegative atom always produces deshielding effect by pulling electron away from the protons.
So, higher the electronegativity, higher the deshielding effect and therefore higher the chemical shift.
For example, in 2-butanol, the two methylene protons are not equivalent and the protons attached to carbon bearing hydroxyl group are more deshielded hence show greater chemical shift.
The location of the electron withdrawing also highly influences the chemical shift values.
For instance, carbonyl group also acts as electron withdrawing group and the protons are deshielded differently based on how they attached with it.
In aldehyde, the proton is directly attached to carbonyl group hence highly deshielded producing a chemical shift within the range of 9 to 12 ppm.
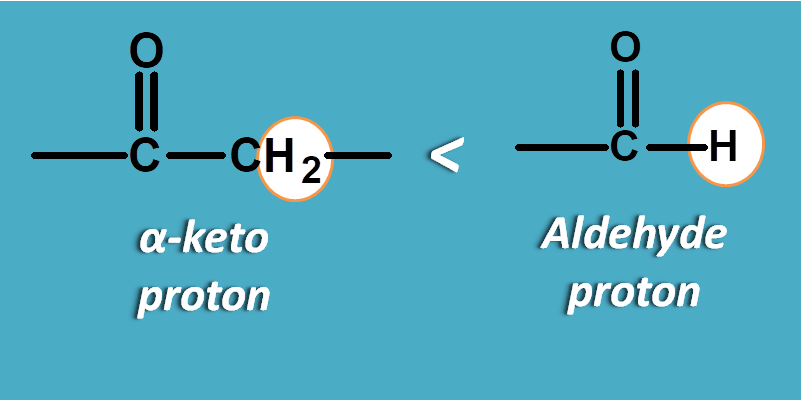
On the other hand, protons attached to the α-position of carbonyl group are comparatively less deshielded hence fall in the range of 1.5 to 2.5 ppm.
You can observe a great variation in the NMR chemical shift values by a small difference in the point of attachment.
2. Magnetic anisotropy
Just above we have seen that chemical shift may vary significantly by the way of attachemetn of protons with an electron withdrawing group.
Interestingly, NMR chemical shifts may also vary with how the protons are arranged within the space i.e. by configuration.
So this concept belongs to the groups having pi electrons which produce their own magnetic field which is variable at different locations.
Hence it’s called as anisotropy - that is non-uniform.
As pi electron cloud lies above and below the plane, the magnetic field produced by these electrons will be in a circular fashion passing through the pi cloud.
Here non-uniformity comes in to the picture
The circular magnetic field will have two opposite directions, one being parallel to the externally applied magnetic field and another being in opposite direction.
The former region is a deshielded region and the later is a shielded region.
So if a proton whether shielded or deshielded, all depends on where it is located, either in shielded or deshielded region.
Let’s take the case of alkenes.
You can observe that magnetic field created by pi electrons is parallel to the applied magnetic field on outside the cloud and opposite inside the cloud passing through the double bond.
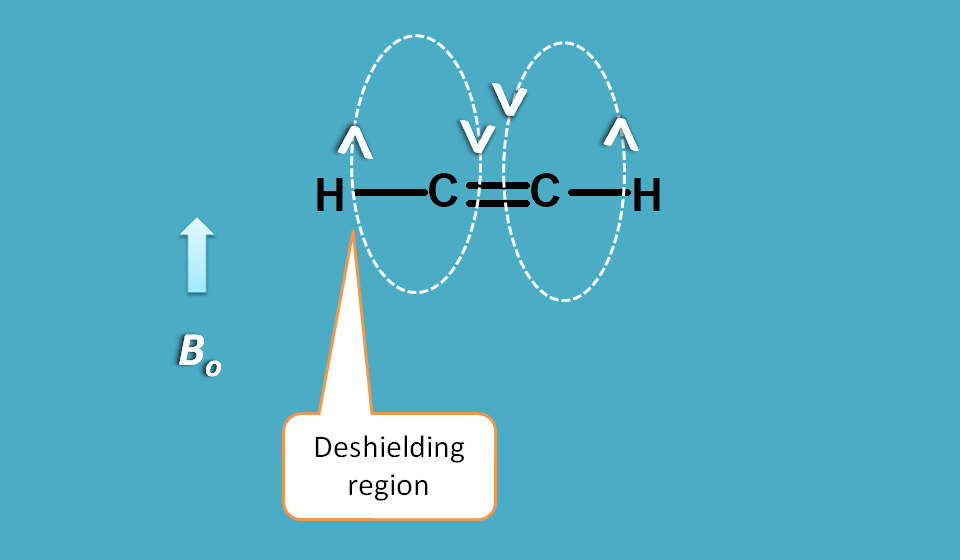
Since protons in alkene fall outside the cloud they are deshielded hence show a greater chemical shift.
Same concept can also be applied for aromatic compounds. These flat compounds again show a pi cloud that is shielding inside the ring and deshielding outside.

So, all the protons are deshielded but this time with stronger pi cloud showing more chemical shift than alkenes.
Aromatic protons fall in the range of 6.5 to 8 ppm where as alkene protons fall in the range of 4.5 to 6.5 ppm.
So that’s about alkenes and aromatic compounds. Then what’s up with alkynes.
Alkynes are somewhat different. They have sp hybridization whereas alkenes and aromatic compounds are sp2 hybridized.
So their structure is linear making a different magnetic field produced by pi cloud this time passing through the protons with opposite direction to the externally applied magnetic field.
Hence the protons are shielded rather than undergoing deshielding. So their chemical shift decreases falling in the region of 2.0 to 3.2 ppm.
Now let’s see another factor influencing chemical shift.
3. Hydrogen bonding
Hydrogen bonding is just similar to the first factor electronegativity except that in this case the effect is shown by two such atoms.
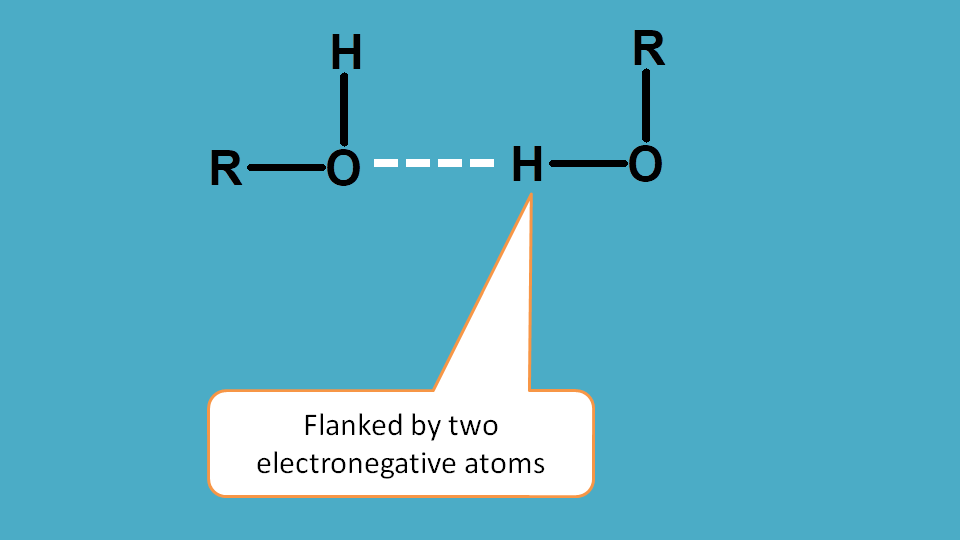
As you know that hydrogen bonding is formed between two electron negative atoms through hydrogen, the later will have very less electron density in its surrounding hence more deshielded.
So protons attached to heteroatom such as halogens, oxygen and nitrogen are deshielded increasing chemical shift.
But, it is to which extent?
This is again questionable.
As you know that the degree of hydrogen bonding depends on the relative concentration of the compound, the extent of deshielding effect is variable among the protons attached to heteroatom.
Hence they don’t produce a sharp peak instead they yield a broad peak with chemical shifts varying to a specific range.
For example, protons attached to alcoholic oxygen can yield a peak ranging from 0.5 to 5 ppm.
So if you observe a broad peak in NMR spectra, we can assume a hydrogen bonding.
That’s about the three factors influencing NMR chemical shifts. This knowledge makes us to predict proton NMR spectra and even assign the groups to the observed values facilitating structural determination.